Dephasing effects in electron transport in molecular systems connected between contacts average out the quantum characteristics of the system, forming a bridge to the classical behavior as the size of the system increases. For the evaluation of the conductance of the molecular systems which have sizes within this boundary domain, it is necessary to include these dephasing effects. These effects can be calculated by using the D'Amato-Pastawski model. However, this method is computationally demanding for large molecular systems since transmission functions for all pairs of atomic orbitals need to be calculated. To overcome this difficulty, we develop an efficient coarse-grained model for the calculation of conductance of molecular junctions including decoherence. By analyzing the relationship between chemical potential and inter-molecular coupling, we find that the chemical potential drops stepwise in the systems with weaker inter-unit coupling. Using this property, an efficient coarse-grained algorithm which can reduce computational costs considerably without losing the accuracy is derived and applied to one-dimensional organic systems as a demonstration. This model can be used for the study of the orientation dependence of conductivity in various phases (amorphous, crystals, and polymers) of large molecular systems such as organic semiconducting materials.
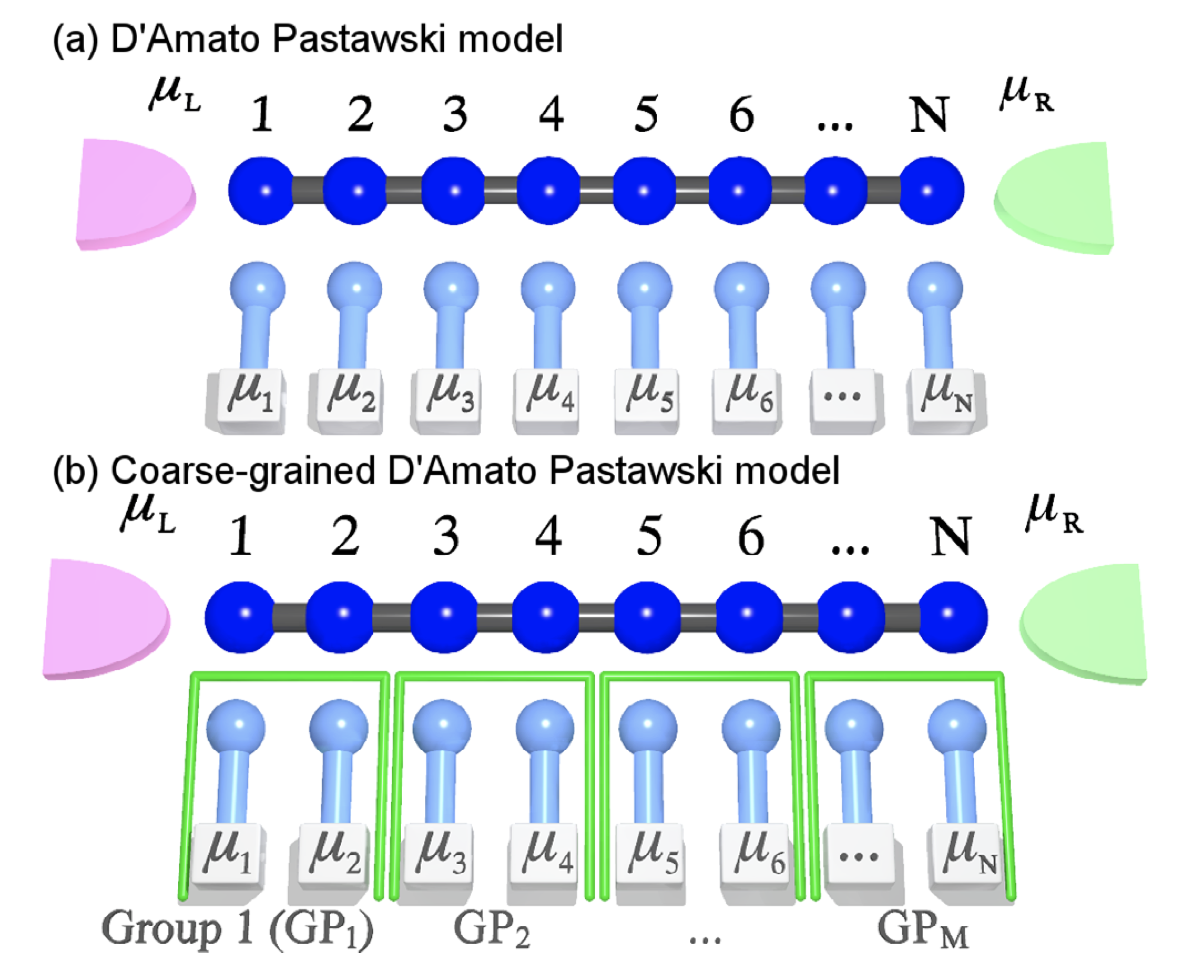

Dephasing effects in electron transport in molecular systems connected between contacts average out the quantum characteristics of the system, forming a bridge to the classical behavior as the size of the system increases. For the evaluation of the conductance of the molecular systems which have sizes within this boundary domain, it is necessary to include these dephasing effects. These effects can be calculated by using the D'Amato-Pastawski model. However, this method is computationally demanding for large molecular systems since transmission functions for all pairs of atomic orbitals need to be calculated. To overcome this difficulty, we develop an efficient coarse-grained model for the calculation of conductance of molecular junctions including decoherence. By analyzing the relationship between chemical potential and inter-molecular coupling, we find that the chemical potential drops stepwise in the systems with weaker inter-unit coupling. Using this property, an efficient coarse-grained algorithm which can reduce computational costs considerably without losing the accuracy is derived and applied to one-dimensional organic systems as a demonstration. This model can be used for the study of the orientation dependence of conductivity in various phases (amorphous, crystals, and polymers) of large molecular systems such as organic semiconducting materials.

